News & information from UB, New York's flagship university
Powerful new imaging method reveals in detail how particles move in solution


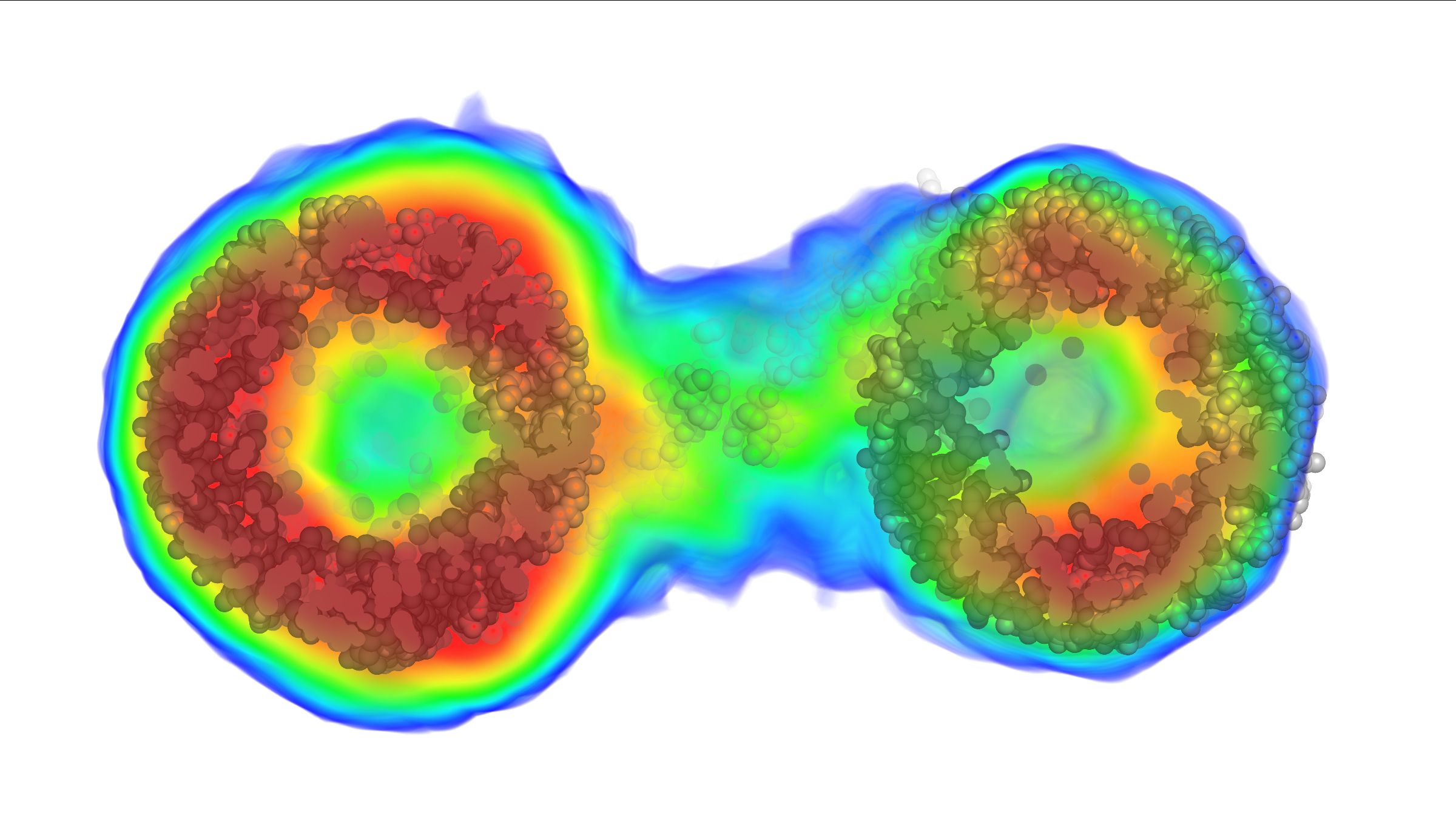
Thanks to the new method, this image of a biomolecule reveals its intricate internal structure in orange, red and yellow. Until now, scientists would only have been able to see the blue outline. (Credit: Image was first published in Nature Methods on Jan. 29, 2018)
X-ray laser method solves phase problem for solution scattering; will improve imaging in areas ranging from astronomy to virology
Release Date: February 27, 2018 This content is archived.


BUFFALO, N.Y. — New research published in Nature Methods will dramatically improve how scientists “see inside” molecular structures in solution, allowing for much more precise ways to image data in various fields, from astronomy to drug discovery.
The new method will allow for the visualization of many more biological molecules, providing critical information about what is inside molecules to scientists who currently can only access their outer shape or envelope. Such information could be a major boost to studies of viruses, for example.
“With existing techniques, you can only see the outline of the virus,” said author Thomas D. Grant, PhD, research assistant professor in the Department of Structural Biology in the Jacobs School of Medicine and Biomedical Sciences at the University at Buffalo and the Department of Materials, Design and Innovation in the UB School of Engineering and Applied Sciences and Hauptman-Woodward Medical Research Institute. “This new method allows us to see inside the virus molecule to understand how the genetic information is arranged, potentially giving new insight into how the virus injects this genetic information into its host.”
Grant is the sole author of the paper, a rarity among papers published in this journal. He is a scientist with BioXFEL (Biology with X-ray Free Electron Lasers), a National Science Foundation Science and Technology Center composed of eight U.S. research universities that is headquartered at UB. Its mission is to address fundamental questions in biology at the molecular level using cutting-edge techniques, including X-ray laser science.
Solving the phase problem
Grant’s method has solved the phase problem for a particular molecular determination technique called solution scattering. The phase problem is where critical information about the phase of a molecule is lost during the experimental process of making a physical measurement.
He explained that most molecular structures today are solved using X-ray crystallography, where the structures scatter intense X-rays in patterns consisting of hundreds of thousands of unique pieces of information, which are used to ultimately reveal the structure at high-resolution.
“The problem is that more than 75 percent of molecular structures do not readily form the ordered crystals that diffract well,” explained Grant. “That means many molecules are difficult to visualize in three dimensions.”
In addition, he said, biological molecules can exhibit dynamic motions that have an impact on how they function but those motions are missing when structures crystallize, resulting in the loss of important biological information.
One way around this obstacle is to use a technique called solution scattering in which X-rays scatter off of molecules floating in solution instead of arranged in a crystal.
“Solution scattering allows the molecules to move dynamically in their natural states, enabling the visualization of large-scale conformational dynamics important for biological function,” said Grant. “However, as the molecules tumble in solution, they scatter the X-rays in many different orientations, losing most of the information, typically yielding only 10 to 20 unique pieces of data.” Until now, such little information only yielded low-resolution outlines of the particle shape.
Grant developed a new algorithm that enables reconstructing the three-dimensional electron density of a molecule, similar to a 3-D reconstruction of the brain produced by a CT scan. However, his algorithm does this using only the one-dimensional data from solution scattering experiments.
Like seeing facial features instead of just a silhouette
“For the first time, this enables us to ‘see inside’ these molecules floating in solution to understand the internal density variations instead of only seeing the outer edges or ‘envelope’ of the particle shape,” Grant said. “Like being able to see all of a person’s facial features instead of just the silhouette of their face, this added information will enable researchers to better understand molecular structures in solution.”
He developed the new method by expanding upon a well-known mathematical technique called “iterative phase retrieval.” This is a computational technique that provides a way to solve the phase problem.
Grant explained: “The phase problem is akin to having a camera that accurately records all the intensities of each pixel, but scrambles where those pixels are, based on a complex mathematical equation. So you're left with a useless image of scrambled pixels.”
Scientists, he said, have typically worked to decode that mathematical equation by changing the image a little bit to make sure it looks approximately as they expect. For example, in a landscape photo, the blue pixels depicting the sky should naturally be at the top.
Solving the phase problem is like decoding that equation, Grant continued, and being able to place all the pixels where they're supposed to be, reconstructing the original image.
“However, this process changes some of the intensities, so you correct them based on the original scrambled image you have,” he said. “This method cycles through this process iteratively, gradually improving the phases with each cycle, ultimately retrieving the final phases, solving the phase problem and reconstructing the desired image.”
Grant’s method, called “iterative structure factor retrieval,” allows scientists to reconstruct not only the three-dimensional phases but also the three-dimensional intensities which are lost in solution scattering experiments as the molecules tumble randomly in solution.
“This is the first demonstration of the ability to reconstruct three-dimensional objects from one-dimensional experimental data and it will likely have a large impact in related imaging fields,” he said.
Media Contact Information
Ellen Goldbaum
News Content Manager
Medicine
Tel: 716-645-4605
goldbaum@buffalo.edu


{kind=link}
{kind=link}