News & information from UB, New York's flagship university
UB researcher narrows time window for administering specific treatment to infants with Krabbe disease

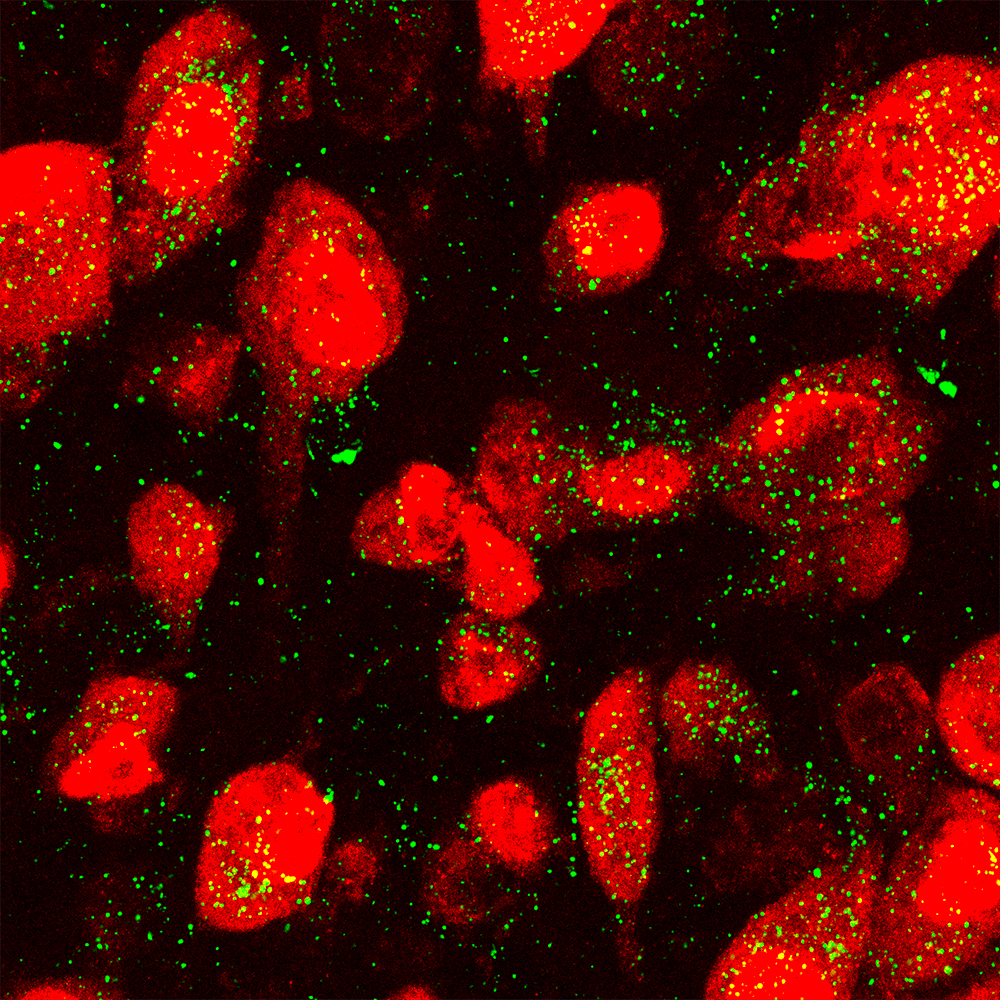
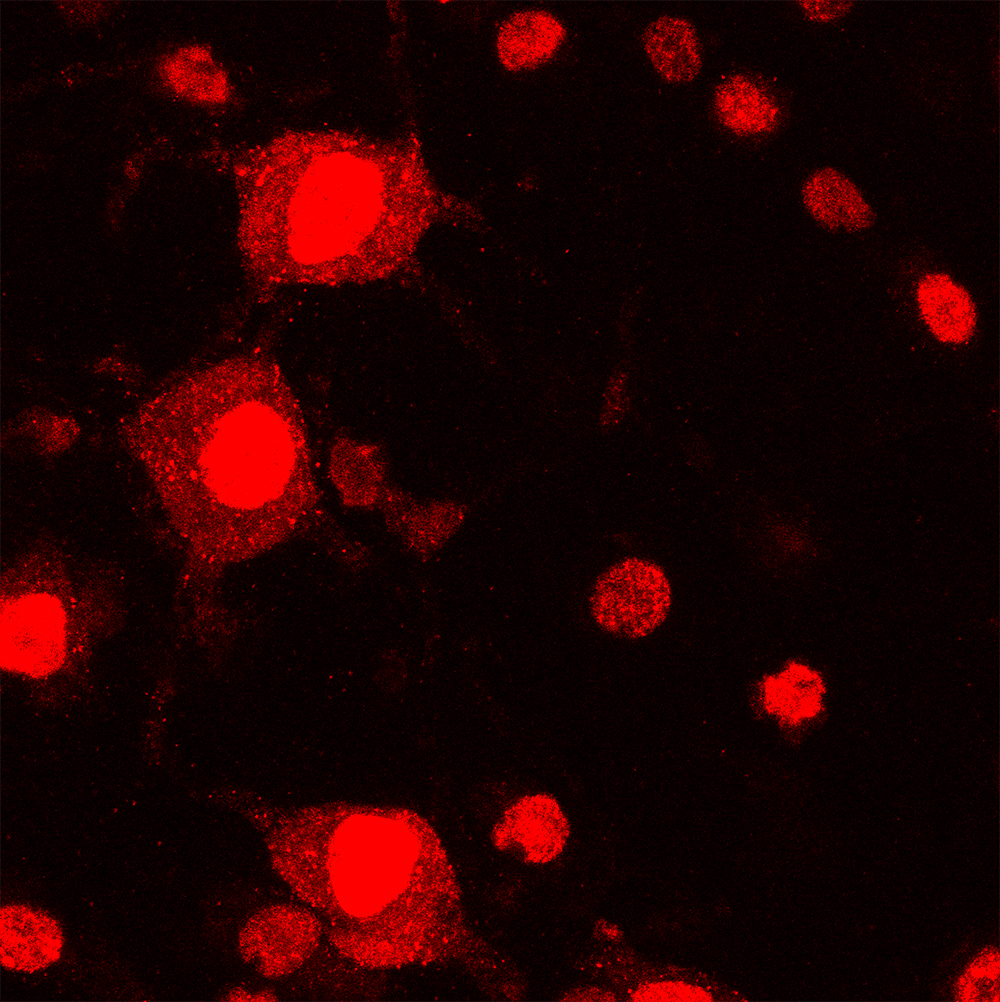
In the confocal microscope image on the left, GALC enzyme (green) is expressed in neurons (red) in the normal mouse brain. In the image on the right, GALC enzyme is absent in the neurons of the Krabbe mouse brain. (Image: Daesung Shin)
Study has implications for other neurological disorders, including multiple sclerosis and Parkinson’s disease
Release Date: November 9, 2020 This content is archived.



BUFFALO, N.Y. – A team of University at Buffalo researchers have published a paper in Nature Communications that is helping to define the best time to give a specific treatment to infants born with Krabbe disease (KD). This treatment has been found to prolong life for these infants, for as long as a few years.
The paper was published online Oct. 23 in Nature Communications.
Daesung Shin, PhD, assistant professor in the Department of Biotechnical and Clinical Laboratory Sciences and the Neuroscience Program, both in the Jacobs School of Medicine and Biomedical Sciences at UB, is the lead investigator of the research. He also conducts research at UB’s Hunter James Kelly Research Institute.
KD is an inherited disorder that destroys myelin, the protective coating of nerve cells in the brain and throughout the nervous system. In most cases, signs and symptoms of Krabbe disease develop in babies before 6 months of age, and the disease usually results in death by age 2. When it develops in older children and adults, the course of the disease can vary greatly.
The progressive neurologic disorder is caused by a deficiency of galactosylceramidase (GALC). GALC is an enzyme that breaks down galactosylceramide, an important component of myelin, which ensures the rapid transmission of nerve impulses.
Although there is no cure for KD, hematopoietic stem cell therapy (HSCT), a therapy that makes blood cells, reduces neurologic deterioration and improves developmental advances. These benefits are dependent on the severity of the disease at the time the stem cells are transplanted, and are only beneficial if delivered at a clinically defined pre-symptomatic time point before symptoms appear.
“Even though it is widely accepted that early treatment is essential for the most positive outcome, the precise therapeutic window for treatment and what happens during this early time have never been elucidated,” Shin said.
To address that issue, Shin’s team used mutations to create a novel mouse model of KD.
“We engineered an inducible knockout mouse for the GALC gene deletion in specific cells at specific times, which provided us with the opportunity to directly ask when and where GALC enzyme is required for brain development,” Shin said.
“We were particularly interested in the role of early developmental GALC function,” he said. “Our study not only revealed a key developmental process that requires GALC in the perinatal period, but also demonstrated that temporal GALC expression is likely a major contributor to brainstem development.”
The researchers found that by increasing GALC levels at or before this newly defined perinatal period they could improve the effectiveness of therapeutic interventions for KD.
“For the first time, our work showed the mechanistic evidence to explain why treatment must occur so early with the defined critical postnatal period at days 4-6 in mice, and demonstrated that temporal GALC expression during this time is a major contributor to brainstem development,” Shin said. “Augmenting GALC levels at or prior to this newly defined period would likely improve the efficacy of therapeutic interventions for Krabbe patients.
“While the time scale between mice and humans is considerably different, the sequence of key events in brain maturation between the two is consistent,” he said. “It was estimated that the mouse nervous system at postnatal days 4-6 corresponds to a gestational age of 32 weeks in humans. Therefore, we anticipate that if our result is correct, then in utero treatments at, or prior to, 32 weeks should have better outcomes than conventional postnatal treatment for Krabbe babies.”
Shin said his team will further identify which cell type needs to be targeted with therapy.
“This work will directly impact the design of novel treatment options for KD patients, he said,” noting that KD studies are at the basis of research on other, more common neurodegenerative diseases, such as multiple sclerosis and Parkinson’s disease. Therefore, the team’s work will have implications beyond KD.
Co-authors on the research were Nadav I. Weinstock, MD-PhD student, and Conlan Kreher, former master’s student, both of the HJKRI and the Department of Biochemistry in the Jacobs School; Jacob Favret, research technician in the Department of Biotechnical and Clinical Laboratory Sciences; Lawrence Wrabetz, MD, and M. Laura Feltri, MD, both co-directors of the HJKRI and members of the departments of Biochemistry and Neurology, as well as the Neuroscience Program.
Duc Nguyen and Ernesto R. Bongarzone of the Department of Anatomy and Cell Biology in the College of Medicine at the University of Illinois at Chicago also participated in the research.
The project was initiated with the support from Empire State development fund for HJKRI, and further developed and finalized by the R01, R56, and R03 grants from National Institutes for Health-National Institute for Neurological Disorders and Stroke awarded to Shin.
The mission of the HJKRI is to unlock the mysteries of myelination in the nervous system and to find therapies for diseases of myelin. The institute was named for Hunter James Kelly, son of the former Buffalo Bills quarterback Jim Kelly. Hunter died of KD in 2005 at age 8. At that time, he was the longest living survivor of infantile KD.
Media Contact Information
Barbara Branning
Media Relations
Tel: 716-645-6969
bbrannin@buffalo.edu


{kind=link}
{kind=link}
{kind=link}